Fibrillin and associated proteins
Fibrillin microfibrils
Among the major components of force-bearing tissues, such as arteries, lungs and ligaments, are the 10-12 nm diameter fibrillin-containing microfibrils. The fibrillins are a group of three proteins, fibrillin-1, -2 and -3, that have primary structures dominated by calcium-binding epidermal growth factor-like domains (cbEGF). Microfibrils have roles not only in maintaining the structural integrity of tissues, but also in the regulation of cytokines through the sequestration of molecules such as transforming growth factor-beta and the bone morphogenetic proteins in the matrix.
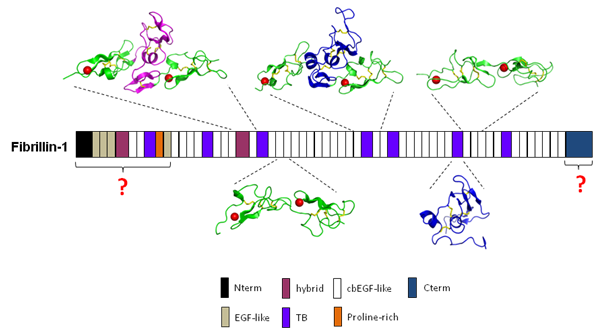
In humans, defects in the fibrillin-1 and fibrillin-2 genes have been linked to diseases that affect the cardiovascular, skeletal and ocular systems, including Marfan syndrome (MFS), congenital contractural arachnodactyly, ectopia lentis, Weill-Marchesani syndrome and stiff skin syndrome. Our research interests include the structure of fibrillin and how it is assembled into microfibrils, the pathogenic mechanisms of microfibril-related diseases, and how microfibrils regulate extracellular matrix growth factors. Our recent studies have identified the molecular architecture of the main module types within fibrillin-1, from which we have proposed a calcium-stabilised extended structure for fibrillin within the microfibril. We are currently studying fibrillin-1 intermolecular interactions with other cell and matrix components in order to gain insight into the processes of microfibril assembly and matrix regulation.
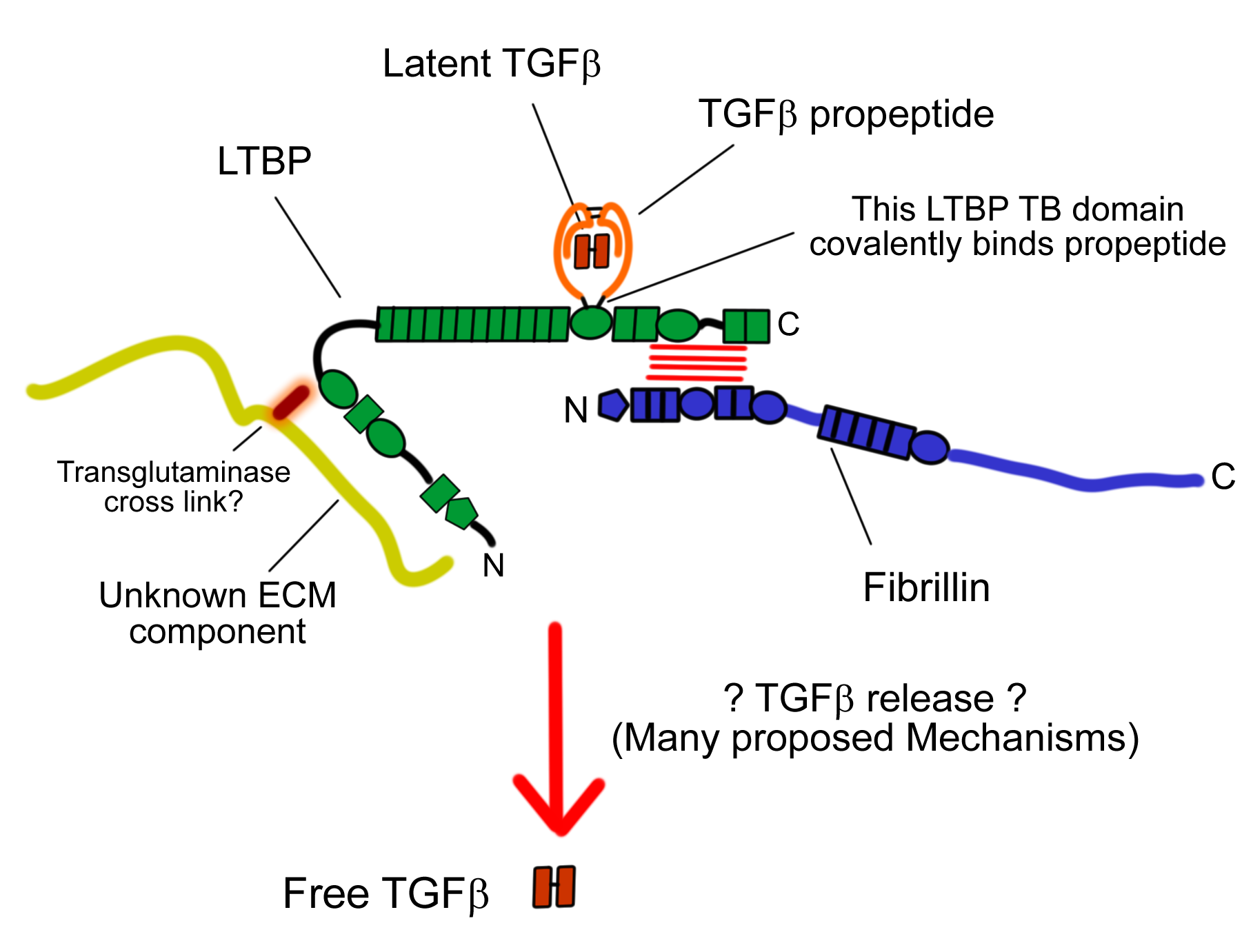
Latent transforming factor-beta binding proteins (LTBPs)
Latent transforming growth factor beta binding proteins (LTBPs) are a class of proteins that have many similarities with the fibrillins. Evidence so far suggests that rather than having a primarily structural function, their main role is in the regulation, targeting and release of transforming growth factor beta (TGF-beta). TGF-beta is an extremely important factor, both in normal development and also in a wide variety of pathological conditions. LTBPs contain many domains in common with fibrillins, as well as tandem arrays of cbEGFs they also contain TB domains, one of which is responsible for binding TGF-beta's pro-peptide through a disulphide exchange mechanism, and in so doing retains TGF-beta in a latent state.
Our aim is to understand how LTBPs function on the molecular level, using structural, biophysical and biochemical techniques in vitro, and with the aim of developing models that can be further tested in vivo by us or our collaborators. Our initial focus is on understanding the interaction between the C-terminus of some LTBPs and the N-terminus of fibrillin. We already have a great deal of experience working with constructs of fibrillin, and this experience has been invaluable in getting to grips with LTBPs.
By understanding precisely how LTBPs are regulated in the extracellular matrix we hope to provide vital insights into the mechanism of TGF-beta signalling, which could have wide reaching consequences for understanding both human development and human disease.
Fibrillin-integrin interactions and Stiff Skin syndrome
Apart from MFS, mutations in fibrillin-1 have recently been associated with Stiff Skin Syndrome (SSS; MIM #184900), which is a congenital autosomal dominant form of scleroderma. FBN1 mutations resulting in substitutions W1570C, C1564S and C1577G within the TB4 domain of fibrillin-1 have been described (Loeys et al., 2010). TB4 is the only domain within fibrillin-1 which contains an RGD sequence that mediates integrin binding.
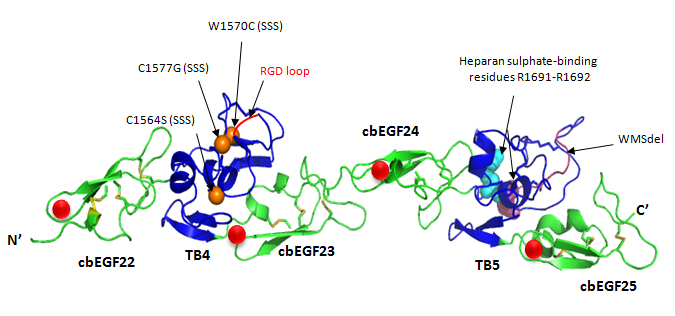
Fibrillin-1 cbEGF22-25 fragment. cbEGF22-25 homology model based on the crystal structure of cbEGF22-TB4-cbEGF23 showing SSS substitutions and WMS deletion. The cbEGF domains are in green and TB domains are in blue. Calcium ions bound to the cbEGF domains are shown as red spheres; disulphide bridges as yellow sticks; integrin-binding RGD motif is shown in red; heparin binding residues as cyan spheres (homology model created using Modeller 9v6 and rendered using PyMol).
The domain-specific nature and unique phenotypic outcome of SSS mutations suggests
a different pathogenic mechanism from that of MFS. In MFS, a "loss-of-function" mechanism
of fibrillin-1 mutations is seen resulting in effects on the cardiovascular, skeletal
or ocular systems. In contrast SSS has no observed effect on these organ systems but
is skin-specific and suggestive of a "gain-of-function" mechanism which results in
over-production of the ECM.
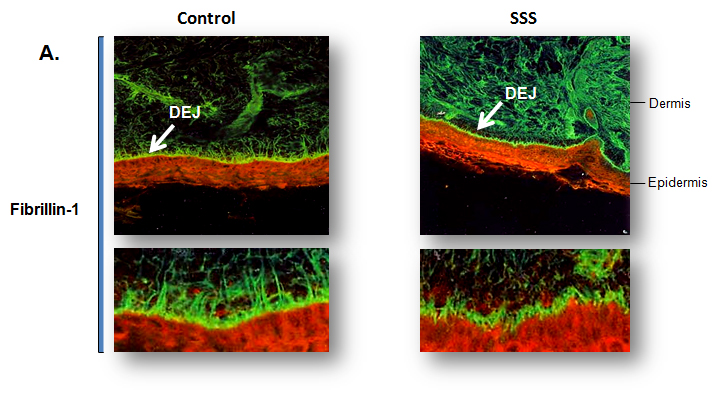
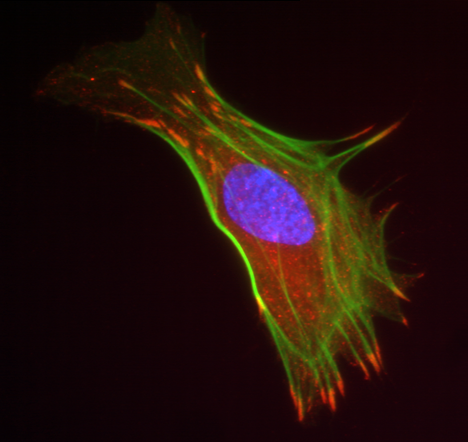
Immunochemical staining for fibrillin-1 microfibrils in control and SSS patient
skin biopsies for fibrillin-1 microfibrils: Immunofluorescence of skin biopsies (10x
magnification) from patient with SSS reveals a dramatic increase in the expression
of fibrillin-1 (green) in the dermis and specifically at the dermal-epidermal junction
(DEJ) when compared to age-matched control. Higher magnification (40x) reveals that
the microfibrillar bundles (composed of fibrillin-1) at the DEJ in patient with SSS
have a stubby appearance without the deep projection into the underlying dermis seen
in the control samples (Loeys et al., 2010).
Fibrosis is a process in which excess fibrous connective tissue forms and is characterised by the accumulation of fibrillar collagens although other ECM components are also over produced. SSS skin shows disorganised macroaggregates of fibrillin-1 that fail to contact neighbouring cells at the dermal-epidermal junction similar to that in Systemic Sclerosis (SSc). A small deletion in the TB5 domain of fibrillin-1 has been found in patients with the autosomal dominant form of Weill-Marchesani Syndrome (WMS; MIM #277600) (Faivre et al., 2003). This domain is upstream of TB4 domain and has been shown to heparan sulphate binding residues which have been shown to modulate integrin binding to the TB4 domain of fibrillin-1 (Bax et al., 2007).The phenotype of WMS is similar to that of SSS patients, short stature, thick skin, joint stiffness, brachydactyly but with the exception of ocular defects involving, anterior dislocation of the ocular lens with severe glaucoma, which is also a feature of MFS. Our group is trying to study the pathogenic mechanisms underlying these various disorders involving fibrillin-1 using biochemical, biophysical and cellular techniques, which will further elucidate its role and organisation into microfibrils in the ECM.